



The DNA Hoax
Part 2: Sequencing

All of the content that is put out on this Substack is going to be for free. If you feel so inclined to donate or Sign up for a Paid Subscription that is very much appreciated. It will keep me writing, putting out content and continuing the largest Control Studies Project falsifying Virology.
THE CLAIM
So here we arrive at the most scary part of the DNA and by extension Virus hoax. Full of big numbers, expensive machinery and highly specific claims of accuracy. This one is going to be a behemoth of a post, there is no way around it unfortunately… gonna leave it all in one article so it is all in one place, sit tight and enjoy.
Please watch the two videos below:
The first being an animation of what is claimed to be occurring at nucleotide level. They obviously can’t show you real pictures of what is happening and have to animate everything because:
A: Nucleotides are so small there aren’t any microscopes in existence that can see them.
B. It isn’t physically happening at all, as this is taking place in a liquid chemical state in solution. All of these strings and blobs and sequences aren’t physically there, they are just proposed hypothetical structures based on electrostatic modelling at the molecular level…. tenuous huh!
This second video shows you the setup, data input and running of the machine itself. Handy to see to orientate yourself with the process. This machine being only a slightly more complex version of a PCR thermocycler: A heating element, a few lasers, a few cameras and a flow cell….. that’s it folks….. One MILLION dollars thank you please!

The Numbers
To fully get your head around what they are claiming, let’s lay out some of the data we will be crunching today:
One ml Saliva sample from a human could contain nearly 500 distinct different microbial species (bacteria, fungi, viruses, protozoa and parasites) with the total count of organisms stretching into the tens of BILLIONS.

Each one of these species of bacteria, for instance the commonly occurring Streptococcus Mutans could contain genetic variations within the same sample of up to 23% of the genome. So tens of billions of organisms, with even the same species 1 of 500 potentially differing by a quarter of the entire genome.

If you thought you could “clean up” this sample and sequence a few of these species, say you were going after unicorns… I mean viruses…. You could apply antibiotics to remove all the bacterial DNA… right!? WRONG… you got the lot in there whether you like it or not.

This is obviously JUST within a saliva sample. If you are sequencing a cell culture, you have the saliva sample on top of the Cell Line on top of the Fetal Bovine Serum.
They take this sample, whizz it up into a big old DNA soup and “cut it” into very small segments, in most cases with Illumina sequencing 150 Base Pairs long and spit them out the other side.
In some of the more advanced Illumina machines they are churning out 10 BILLION reads, 150bp long.

Context summary for the sample being tested:
The inputs going into their machine are supposedly 10 BILLION short little segments that it is not known from which of the Billions of microbes they came from, all floating around in liquid chemical state in a solution so not physically there, merely hypothetically modeled to be in a sequence.
THE INPUTS
Going into this we are going to start with two massive assumptions that A: DNA and Nucleotides exist and can form these sequences and B: They can be read by an Illumina Sequencer. This is certainly not my opinion as they both come with an equally sized to this article, litany of logical flaws and methodological errors. But for sake of brevity we will continue on starting with these assumptions as a base.

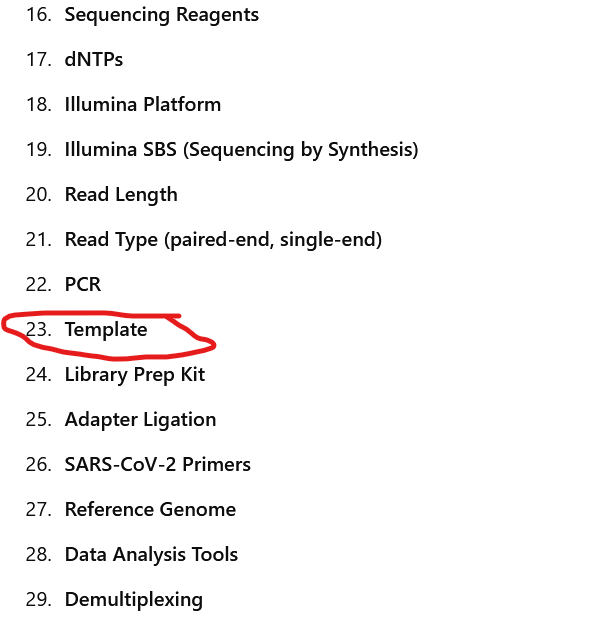
Here we have all of the input variables going into Illumina Sequencing. I will deal just with Illumina Sequencing again to keep it brief. Nanopore Sequencing is almost identical in its input variables , it is much less accurate than Illumina sequencing giving back 15% higher error rates because it *attempts* through the same problematic constructions to give longer reads.
Sanger Sequencing is just a glorified version of PCR, targeting only specific sections of a genome with pre-made Oligonucleotides sequences, so the sequence MUST be known BEFORE to target something (not that this stops them claiming to be able to De Novo sequence with it (Quacks)).
Not a single one of these input variables has ever been controlled for at any stage by anyone. I.e do not Reverse Transcribe a sample and keep all other chosen variables the same to see the effect it has on the outcome.

This obviously renders the entire process of sequencing redundant as a Pseudoscience, but you know me… we are going to pull it apart.
HYPOTHESIS
The intentional choice surrounding the reagents and methods of the 29 distinct steps of sequencing is a determining factor over the sequences generated.
CHOOSING OUTCOMES
Circled in red is the Template otherwise known as the sample. Out of the nearly thirty distinct methodological procedures, some of which contain tens to hundreds of reagents (liquid chemicals) and methodological steps in their own right, ONLY the template is (partially) a “natural” input variable.
The template is only partially natural due to the many choices intentionally made depending on what someone *thinks* is in the sample. Hopefully by the end of this you will truly see how the choosing of inputs sculpts the output sequence.
If a clinical sample is thought to contain a virus, it will be put into Viral Transport Medium to “preserve” for sequencing.


It actually doesn’t preserve the cells at all as it is in 2% FBS and Nephrotoxic Antibiotics which we have shown to cause “CPE” in cell lines. Also Fetal Bovine Serum is unwittingly introducing contamination with a source of bovine genetics. Is this factored in ( I highly doubt it).
RNA ONLY
Right from the off, the moment someone has decided a sample contains a virus it is immediately treated in a different way with the type of reagents used. We then go into Reverse Transcription. Because someone *thinks* a sample contains Sars Cov 2 for instance, which is an RNA virus, this has to be reverse transcribed otherwise the Illumina Sequencer won’t be able to even sequence it.

This Reverse Transcription has it’s own quite long methodological procedure involving not only a complete set of specific reagents even to extract RNA that differs from our well known Soapy/Salty DNA extraction.

Our Reverse Transcription ALSO contains a primer set that can be specific to a sequence that you want to look for in a sample .i.e have assumed it is there.


Before our sample that someone assumes contains a virus even gets near the sequencing machine it is “amplified” using PCR. Specific Oligonucleotides are synthesized to supposedly amplify all of the genetic material of Sars Cov 2. Again this is entirely uncontrolled and the sheer fact they are putting in KNOWN synthetic sequences and then lo and behold FINDING those sequences when sequencing is… well …pretty unsurprising!!


This snaps into focus one of the largest problematic areas being, how did someone carry out the first sequence of any virus if its genome had to be amplified and targeted to “find it”. Like was it just a very fucking lucky guess? Lol.. of course not… it is all circular reasoned, benchmarked and verified against itself.
None more so evident than with Sars Cov 2. They used the “Takara Protocol” a particular set of primers, library prep, adapters and Poly T adapters to TARGET specific parts of a genome ……. that they didn’t know!!!!! How was this specific sequence targeted with oligonucleotides that require you to KNOW the sequence…. the mind boggles!!

Gel Electrophoresis
Next up on our list of determining inputs is the rather innocuous sounding “Fragmentation and Size Selection”. This is the “cutting” up of the DNA strands with enzymes and then putting them through Gel Electrophoresis claimed to be separating out different “sizes” and therefore “lengths” of DNA into bands. This “technique” is claimed to be so sensitive it can give you exact lengths of purified DNA strands. In the case of “Sars Cov 2” Illumina sequencing they determine this number to be 150bp, a chosen number to best “amplify its regions”…

I am going to concentrate on Gel Electrophoresis because it is basically how EVERYTHING in virology is verified. Actually when you dig down into the literature, in oligo synthesis, enzyme synthesis, protein synthesis, Amino Acid Synthesis, Antibody synthesis, peptide synthesis etc etc… In fact ALL of the moving parts that are called “biochemical” molecules and how they are Identified and hence how they are synthesized to be SPECIFIC to the processes of PCR and Sequencing are totally predicted on various forms of gel electrophoresis.
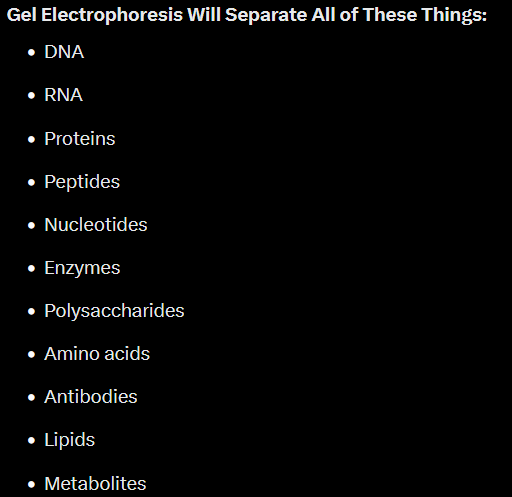
The principal is exceptionally simple; Take a DC battery, a gel a bit like agar-agar, put the cathode at one end of the gel and the anode at the other… Because DNA is supposedly negatively charged it moves through the gel toward the anode and is “filtered out” by the pores in the gel. This “separation” is not to do with charge as the charge difference between size of molecules is so minuscule it doesn’t affect the distance traveled, which ultimately causes the banding, more than the determining factor of FRICTION. Yes this entire principle and hence the entire “specificity” of PCR and Sequencing is based on FRICTION of molecules through a jelly.

Above is all of the “biochemical” molecules that are claimed to work in Gel Electrophoresis… They ALL have negative charge, they are ALL so small they have never been photographed or seen as no microscope exists that will image things this small. Apart from the the lipids and polysaccharides they are essentially all identical in rough function and supposed size. Are they really distinct separate things or just assumed categories with made up functions for what is essentially just negative charge…. my thoughts firmly rest with the latter.
Here is a short video showing you the method and equipment.
Here is a pretty long and boring video on CHOOSING the right Gel, this is important because remember it is all about FRICTION and the TINIEST changes in concentration of the gel causes completely different results. Interestingly Proteins are most commonly separated with a Polyacrylamide Gel… BUT if you are looking for 150bp length DNA you also use the EXACT same equipment including the same gel… something to consider. But nevertheless give it a watch to truly see for yourself the sheer quackery and inaccuracy clearly present in this “technique”.

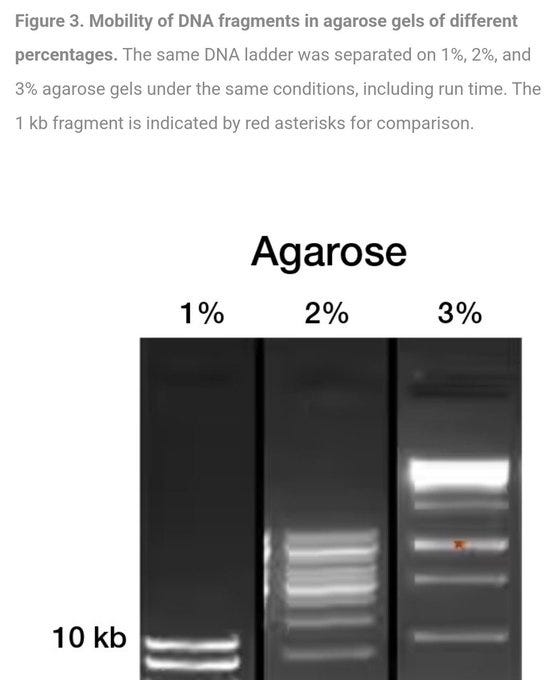
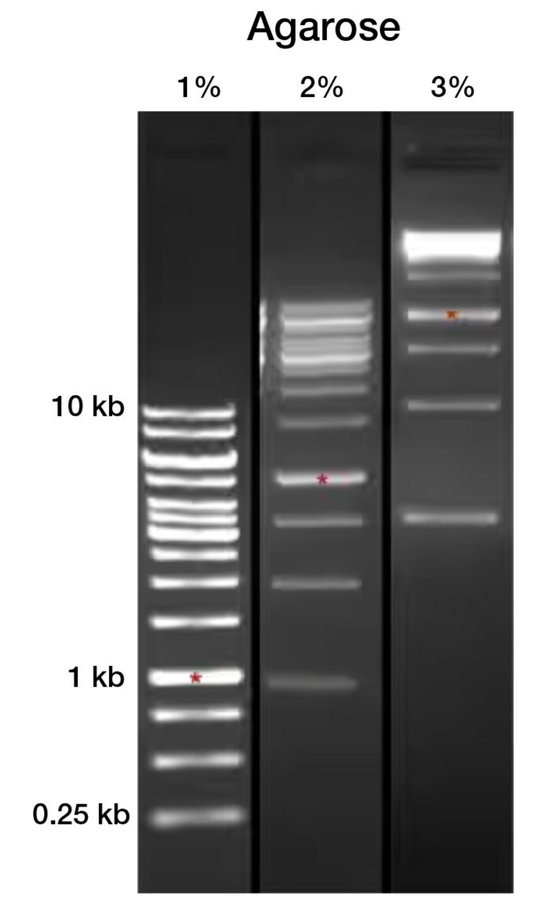
Here are three gels differing by a mere one percent in Gel Concentration mix with the SAME sample… Look at the HUGE difference in results… clearly completely different… This is assuming the gels are consistent and uniform across the entire tray…. which inevitably will be incredibly hard to prove.
PCR and Sequencing Specificity
The fundamental principal of PCR and by extension Sequencing specificity is that an Enzyme, usually Taq Polymerase can “recognize” where a Primer finishes so that the enzyme can then “build” the rest of the nucleotides strand and “copy and amplify” the code… Keeping in mind once again ( a running theme) that this is all in chemical liquid state, not actually physically happening.
This Enzyme however is NOT specific to the Primer sequence… largely because they claim it can do too many things for it to be chemically possible to be specific…

So…. this blob of protein (negatively charged thing identified and verified in a gel using the above technique) is NOT specific but somehow can “recognize” a primer (chemical liquid), not chemically BIND to it… just “interact” with it based on electrostatic charge “structure” (which isn’t actually a physical structure just a hypothetical modeled one in chemical liquid state)…. Tenuous? Nah mate.. not even started yet.
This Enzyme can recognize where to start its “activity” because it recognizes a Primer-Template hybrid. Unfortunately however… this primer-template hybrid containing the SPECIFIC REGION TO BE AMPLIFIED is ABSOLUTELY IDENTICAL in chemistry to a normal Template-Template hybrid which could be any DNA fragment in the sample. They try and claim the IDENTICAL CHEMISTRY …. one is “available” and one is not… despite them both being “free” and of the same Hydroxyl group…

How is this specificity confirmed? I here you cry….. well you guessed it by connecting a DC car battery ( jokes- but it is no much more sophisticated than an AC/DC power supply with a variac on it) into a Jello. This is seen here in Kary Mullis’ paper claiming that because some bands get thicker when he changes some stuff, that means his enzyme (which he verified in his car battery jello machine) is recognizing his specific/nonspecific/identical/available/non available primer(which he verified in his car battery jello machine) …
Here’s the paper for a laugh: MULLISYOUFRAUD
So back on with the Sequencing show. They put this pre-amplfied material through a gel. Assume one of the bands contains all of the same 150bp lengths of DNA segments and then physically cut that piece of gel out….
VOILA… OUR SAMPLE IS NOW READY TO SEQUENCE
Prepping the Library
If I could draw your attention back to the video at the top of the article of the lovely Illumina representative showing us how to perform a run on her machine.
Notice the things that are programmed into the computer. She puts the exact Read length. She then chooses from a drop down menu her Library Prep and Indexing options. She then turns to camera and adds “this includes custom library prep kits”.
The Library Prep kit is essentially all of the reagents that go into sequencing the sample, we have already covered the RNA extraction, The Reverse Transcription, the fragmentation, size selection and PCR amplification. These are all part of the Library Prep. All that is left if the Sequencing Primers and the Adapters.
So as mentioned before with a very targeted sequencing such as that of “Sars Cov 2” the library kit they choose is called the “ARTIC Protocol” covering all of these reagents. This would fall under the category of a “custom library prep”. But even so in what is considered “standard” Library Prep kits available to CHOOSE what you *think* is in the sample to sequence some of the most common ones get as specific as single celled RNA sequencing with SMART-Seq.
Here really is a large part of what I would call “crafting” or “sculpting”. They are knowingly giving quite a bit of information and feeding it into the sequencer, telling it how many BP the reads are, exactly what it is LOOKING FOR… i.e RNA and Small and Single Celled.


ARTIC PROTOCOL
Let’s snap in to the ARTIC protocol for sequencing Sars Cov 2. It contains a Primer SET of more than one hundred different synthesized oligonuleotides that cover the WHOLE genome on Amplicons. So for this particular library prep they are intentionally putting in a large portion of the sequence, to find the sequence… I mean, is it any surprise that you find the thing that you put in there??!!
Lest I just jog your memory from the initial part of the methodology that this sample has ALREADY been amplified by PCR intentionally putting in the sequences they are looking for on top of the potentially specific primer set for Reverse Transcription… to then have another 120 Primer sequences chucked in there for good measure.

Adapters
As If there weren’t enough synthesized oligos in this brew already with all of the primers, they make some more for good measure. The Adapter is chemically bonded onto the flow cell where the LIQUID CHEMICALS (going to get tired of pointing this out) flow over. These supposedly dangle like seaweed ( if it were physically there but it isn’t) and “catch” the DNA going past. It is the front of house “primer sequence”.
I will caveat all of this with the fact that these are trimmed i.e cut out at the alignment phase as they will appear at the front of every read. They are not technically primers because they are supposedly not present in the genome of the thing you are sequencing…
Strange then that these Adapters can be CUSTOMIZED to “catch” single celled RNA.

FLOW CELLS and READ LENGTH
From the start of the protocol and the electrophoresis part we have already customized the number of Base Pairs in each read, this is then programmed into the sequencer so that it “knows” how long the reads are meant to be. The Flow Cell which is essentially the reaction plate where the magic takes place, the particular variety of flow cell is then chosen based on the exact read length…

The big zinger here is that there could have been some sort of verification that the stuff they cut out of the band in electrophoresis was indeed as long a chain as they thought it was (huge amounts of assumptions noted)…. But NO, each cluster does not represent one read… it is essentially just the computer algorithm within the sequencer which chops up the reads into the desired length.. in our case 150bp

P5/P7 Primers
WAIT! You thought we had finished with the amount of primers used! How naive! Nope we have some more, totally synthesized, totally specific sequences intentionally put into the mix. These are specific primers that can match the specific Adapters to “catch” more of the specific DNA around the clusters where they are attached to the Flow Cell…
I promise that is all for the primers.

AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA
OK If there is one piece of information about this entire sham of a technique that adequately shows the sheer level of stupidity involved, it has to be the Poly A Tail.
When sequencing RNA viruses, presumably the first time they did it, they found that somewhere in the middle of this sequence there was a string of Adenine groups all in a row. 20 to 200 (yes TWO HUNDRED) in a row AAAAAAAAAAAAAAAAAAAAA…..

Now instead of just binning this obvious computing glitch (what is the likelihood of 200 Adenine groups in a row?) they decided to start calling it a feature of RNA viruses called the Poly A tail. Now do you think that they could just replicate these findings with no added changes? HAHA… could they FUCK.
They synthesize an almost exact length of Thymine groups and PUT IT IN THE MIX TOO!!! Called the Poly T Adapter they literally make and insert the feature to find it…

ALIGNMENT
The flashing lights display from the fluorescent dyes they purposefully put in there has occurred and this has been spat out as some letters in the predefined length of 150 Base Pair Reads. There are up to 10 BILLION of these with the high end Illumina sequencers.
Now I am not going to bother to go into the inherent flaws within the actual running of this process of which there is a VERY long list. They are mostly revolving around errors in reading and interpreting the order of the lights, which in and of itself is quite a big problem.
Essentially most of these lead to just degradation of the data and I am not particularly interested in these inaccuracies. Just like the high cycle threshold claim of inaccuracies with PCR, whilst legitimate, is rather superficial and doesn’t touch any of the mountain of logical flaws/impossibilities and “sculpting” of outputs that we are covering here.
Nevertheless here is some areas to investigate if this is of interest.
This part of the process could most certainly be an article in itself as it is the part of the method that contains the most obvious ability to figuratively generate fraudulent data. This is not, by the way, my opinion by any stretch of the imagination, this is the admission of any intellectual honest geneticist and certainly the opensource software alignment programs maker's’ like MEGAHIT.
The premise is fairly simple. You take the reads, in our case 150 Base Pairs long and you try and “stitch” them together into contigs to then match them against a database of known genomes. The two biggest problems are A: How this is assembled and B: The genomes they are being referenced against have ALL been built with the logical flaws and “sculpting” of inputs i.e Circular Reasoning (referencing against itself- read my article entitled Benchmarking Reality for more).
Whilst the mathematics that goes on under the hood of these software programmes is quite complex and certainly data heavy, the terminology and basic principles are quite easy to understand. Reads of 150 Base Pairs are split in Kmers which are at the start and end of the reads. These have a CHOSEN value, usually around 21. This represents an “Overlap”. So the computer programmes try to find these Kmers that overlap reads. They join the reads that have an overlap to form a Contiguous sequence (Contig). The Contigs represent a genome and can be compared to the genome database to “identify” something in the sample.
The real bread and butter of the manipulation occurs in these Kmer sections as these computer programmes employ a thing called the De Bruijn Graph method of alignment. This takes the sequence within the Kmer and slides the sequences around to try and force the reads to overlap.
The very fact that these computer programmes are heavily manipulating the overlaps means that THESE CONTIGS ARE NOT NATURAL. As soon as there is an admission that these sequences in ANY way are artificial that means they CANNOT be used to identify anything in nature. End of story.


This is a short but very good video demonstrating how these artificial contigs are generated. You can try this program out for yourself here : De Bruijn Graph
Again for the sake of this article becoming book length, purely because of the sheer amount of problems with all of these methods I will just list all of the problems that knowingly arise with the De Bruijn Graph assembly that give artificial results.

No more is this alignment process shown as being fabricated nonsense than if you start to compare the same datasets using different alignment programs as they did here in this Published paper.

They performed a whopping 6648 De Novo assemblies with all of the well known assembly programs.

Despite the HUGE number of attempts they had they had to admit that in MOST cases they were not able to "assemble" I.e replicate the previously known (invented) genome.

The number of Base Pairs in contiguous alignment varied Massively for 29k right the way down to 1100.

The total fraction of the genome found, varied by TEN FOLD across different assembly programs
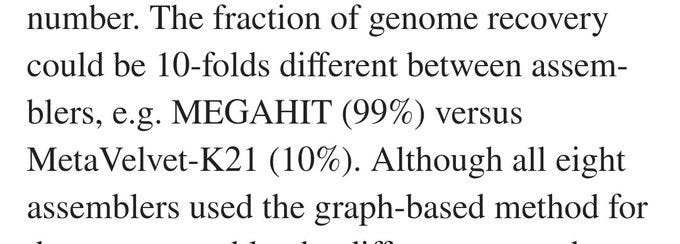
So given this, it is probably no surprise that when sequencing Sars Cov 2 with Nanopore that more than half of the assembly is errors or manipulations.
I guess now the threshold for accurately assembling a genome is just 50% coverage… noted.
FORCING THE WRONG CONTIGS
Whilst learning about the process of sequencing and alignment, one piece of faulty logic stuck out like a sore thumb to me. The implications of this means that not only do these sequences not occur in nature but they can’t possibly be from a single organism, in fact the contigs aligned in the method currently used specifically FORCE the WRONG reads together.
This is easiest to explain if we take a simplified best case scenario, where complete single contigs from one distinct biological entity are sequenced from start to finish splitting them into short reads.

In this instance, quite clearly the reads from one distinct biological source of the DNA should NOT OVERLAP.
Now here it states and I have had this corroborated numerous times that the sequencer DOES NOT know the Kmer value used in alignment to find Overlaps input into it. I.e If the sequencer knew the Kmer value when sequencing and went forward and backward like a bleed margin by the Kmer value, you could then stitch the contigs together 100% of the time with 0% errors.

Considering the FACT that NONE of the alignment programs work this way, they MUST be fabricating a sequence not found in nature.



BOOOOOOM Through pushing Chat GPT it finally turned over its hand to agree that it is a statistical certainty that the sequences generated are fabricated and therefore there is no proof they exist in nature.
I made this quick graphic to try and illustrate the point more clearly.
Genome 1 and 2 represent the complete contigs (60 Base Pairs) of 2 distinct biological entities, lets call them bacteria, lol. The different colours of sequences represent the Reads (20bp) The Yellow section represents the Kmer (5 bp).
So in our first example, just sequencing our bacteria using a best case, pure example of having the genome of one individual bacteria. Note the boxes in red represent an attempt at an overlap using the De Bruijn Graph method. In this case the Original sequence of the bacteria WILL NOT be assembled as there is clearly no overlap.
In our second example introducing a second bacteria. Here we can clearly see that if using the De Bruijn graph method that not only will neither the first nor second bacteria will be aligned in order, but because the first and second bacteria share an exact or similar kmer , they will be forced into a completely fictitious contig.

Here we have proved that this method of alignment CANNOT sequence a single celled organism on it own. The rescue device that is employed is that because there are numerous of these micro organisms that when a kmer is found to match between two reads that this must be representative of a “species” of micro-organism.
If this were the case then there should be little need to manipulate the reads with the De Bruijn Graph method. If they exist in such high quantities i.e Billions of Micro-organisms in a single sample, 10 Billion short reads, you should expect to find alignment easily. Yet chat GPT agrees it would be “Very Hard” to make any contigs if these reads weren’t manipulated. No Shit.

If one used a high Kmer value and no De Bruijn Graph manipulation it would be virtually impossible to make ANY contigs at all.

CONCLUSION
When dealing with specifically “viral” sequencing, which is the main reason why we are here, certainly RNA viruses which are 95% of supposed pandemic causing, I think I have adequately shown that every single minute step of the method is synthesized, manipulated and chosen to sculpt the output.
Certainly in the case of the Primers, literally putting in the sequences they are supposedly looking for, not once but FOUR times with HUGE genome coverage, they are clearly CREATING the sequences they are LOOKING for. None more so obvious than with the Poly A Tail/Poly T tail Adapters.
Some of the evidence that would add weight to this is the fact that if you do not carry out just ONE of these steps being Reverse Transcription, you could apparently NOT be able to sequence even a pure sample of Sars Cov 2 if it were not.


Just to show you THEIR explanation for supposedly why… RNA and DNA are IDENTICAL in chemistry apart from one oxygen extra on RNA. Now all of the binding happens to the Hydroxyl group (OH) which BOTH RNA and DNA have… but somehow an enzyme can Electrostaticly (The chemistry is identical and doesn’t react just “Interact”) “SENSE” the difference of its “structure” (not that it is physically there as it is a chemical in a liquid state). How do they know this? You guessed it… verified by our beloved Car Battery and Jello method…(TBC).
Here we have it, the extremely tenuous excuses as to why the sequences are only produced if they try and produce them. This is a clear decision making process that Sculpts the output through numerous choices made based on what a geneticists THINKS is in the sample.
Really, not even the geneticists are confident in the alignment process as even the mainstream consensus is that there is an “error rate” and list of problems as long as your arm. The real damning evidence rests with the fundamental principle of stitching small sequences together that A: Cannot possibly be from the same Micro- organism and B: Cannot be considered Natural as the De Bruijn Graph method knowingly fabricates sequences.
If Genetic Sequencing CANNOT show that a sequence is present in nature then it CANNOT be used either by itself or PCR to show that someone has any Micro-organism in them, causing an infection or otherwise, especially a “virus”.
HOW TO TEST THE HYPOTHESIS
I have laid out in this article the myriad variables that serve as sculpting inputs into the sausage machine. Considering a high read count Illumina run is almost $2000 per sample, if we were to try and control for every single variable in this method, I have no doubt that the test would be creeping close to the million dollar mark. I think this approach, whilst giving us absolute proof as to which of these variables is having the most influence on outputs, is wholly unnecessary.
To me, I think it is clear the areas that could potentially have the most influence, that being the Reverse Transcription, PCR amplification, the Library Prep and the Alignment. These are the variables that we have chosen to try and control. The Alignment is much more in our control given that part of the project is someone very capable of handling the Alignment programs and data sets.
When conducting our controls we use blinded independent and accredited Contract Research Organizations (CROs). So the way we have tried to get them to alter these input variables is by commissioning them to LOOK for specific things in the dish. For our negative control we have told them just to DNA sequence a cell line and they have treated it one way. For the first of our test cultures we have told them we have found a “virus” in our culture due to CPE present from a clinical sample and want it RNA sequenced to sequence this specific virus.
This is the blueprint of the way I think it is best to control this methodology if any others are interested in conducting their own studies.
Watch This Space.

OMG, I dont have your skill set of what you just presented, but I certainly have the brain cells to understand this is the most insane made up shit I’ve ever seen. These people truly make everything up. The Illumi woman is creepy AF. Like watching a horror film
Whoa, this ain't an easy bake oven.
It's a super complicated one.
Reminds me of the crazy gadgets that the "wizard"of oz had.
No wonder why it's a known defense to explain in court that a DNA match is not reliable 😂.... It's fisher price, not fisher thermo 😆